Maldi-TOF Analysis

AxisPharm Laboratories is equipped with a Voyager DE MALDI-TOF system made by Applied Biosystems. We are routinely using this instrument in analyzing polymers, oligonucleotides, peptides, proteins, and many other macro-molecules that would be difficult or not possible to be detected with API sources or any other mass spec techniques. Masses of peptides (low molecular weight, 750-4,500) can be determined on low picomol quantities with an average mass accuracy about 0.2%. Under optimum conditions, the limit of sensitivity of tryptic peptides (below 4,000) is in the lower femtomol range. Masses can potentially be obtained on numerous biopolymers including oligosaccharides, nucleotides and proteins that range from ~600 to 750,000 Daltons.
During sample analysis on MALDI-TOF, the sample is dissolved in a volatile solvent, and then spotted onto a sample plate with a total volume up to 2 µl. Depending on the type of the sample, a matrix may be spotted before, after, or mixed with the sample on to the same spot where the sample is. Spectral data acquisition takes place after the sample is completely dry and placed inside the high vacuum chamber of the instrument. Salts in the sample can seriously affect the data quality, many times totally suppress the ionization of the sample. Therefore, it is critical to reduce the concentration of any light metal salts in the sample before performing MALDI-TOF analysis with it.
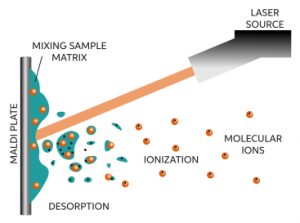
So far the mechanism of MALDI ionization is not entirely clear yet. However, its configuration is fairly simple. It strikes a UV laser (337 nm) on to a dry sample deposit together with a matrix on a metal surface to generate ions from high mass, non-volatile samples such as peptides and proteins. The key to this technique is that in the presence of an aromatic matrix large molecules like peptides ionize instead of decomposing. The process may involve absorption of UV light by the matrix followed by transfer of this energy to the peptide – which then ionizes into the gas phase as a result of the relatively large amount of energy absorbed. To accelerate the resulting ions into a flight-tube in the mass spectrometer they are subjected to a high electrical field.
Three different models have been proposed to explain desorption of the matrix-sample material from the crystal surface: (1) quasithermal evaporation as a result of increased molecular motion, (2) expulsion of upper lattice layers, and (3) an increase in the hydrodynamic pressure due to the rapidly expanding molecules in the crystal lattice. However, there is no consensus yet as to how the sample molecules are ionized. The widely accepted view is that, following there desorption as neutrals, the sample molecules are ionized by acid-base proton transfer reactions with the protonated matrix ions in a dense phase just above the surface of the matrix. The protonated matrix molecules are generated by a series of photochemical reactions.
The matrix performs two important functions: (1) it absorbs photon energy from the laser beam and transfers it into excitation energy of the solid system, and (2) it serves as a solvent for the analyte, so that the intermolecular forces are reduced and aggregation of the analyte molecules is held to a minimum. Some desirable characteristics of a typical MALDI matrix are:
- A strong light absorption property at the wavelength of the laser flux
- The ability to form micro-crystals with the sample
- A low sublimation temperature, which facilitates the formation of an instantaneous high-pressure plume of matrix-sample material during the laser pulse duration
- The participation in some kind of a photochemical reaction, so that the sample molecules can be ionized with high yields
Several matrix-laser combinations have been tested successfully. For peptides and small molecular mass proteins (<10,000 Da), good results are obtained with a-cyano-4-hydroxycinnamic acid (CHCA), whereas high-mass proteins are analyzed with sinapinic acid. The use of 3-amino-4-hydroxybenzoic acid and 2,5-dihydroxybenzoic acid (DHB) has been recommended for the analysis of oligosaccharides.
Our MALDI instrument is a linear model with one meter flight tube length. The ions travel down a linear flight path and their mass/charge (m/z) ratio (see below for an explanation of the difference between mass and mass/charge ratio) is determined by the time it takes for them to reach the detector. Hence, this instrument is called a time of flight (TOF) instrument. The relationship that allows the m/z ratio to be determined is E = ½ (m/z)v2. In this equation, E is the energy imparted on the charged ions as a result of the voltage that is applied by the instrument and v is the velocity of the ions down the flight path. Because all of the ions are exposed to the same electric field, all similarly charged ions will have similar energies. Therefore, based on the above equation, ions that have larger mass must have lower velocities and hence will require longer times to reach the detector, thus forming the basis for m/z determination by a mass spectrometer equipped with a time of flight detector.
During high voltage extraction of the peptide ions produced by exposure to UV light, there are slight differences in the amount of energy that is actually acquired by similarly charged ions. In a linear instrument, these differences result in slight differences in times of flight which results in broader peaks and lower mass accuracy. In terms of resolving fragment ions, a reflector also compensates for similarly charged ions having slightly different overall energies (the more energetic ions that have slightly faster velocities will penetrate further into the ion mirror and hence be slightly delayed relative to less energetic ions – thus both will tend to reach the detector at the same time). As a result, the reflector improves both resolution and mass accuracy. Although there is always the possibility of observing fragmentation ions when using the reflector (and mistaking these for contaminating peptide ions), by adjusting the settings on the instrument it is possible to minimize the possibility of seeing peptide fragmentation in the reflector mode.
Send Request
Other test types:
Related Polymer PEG Products: